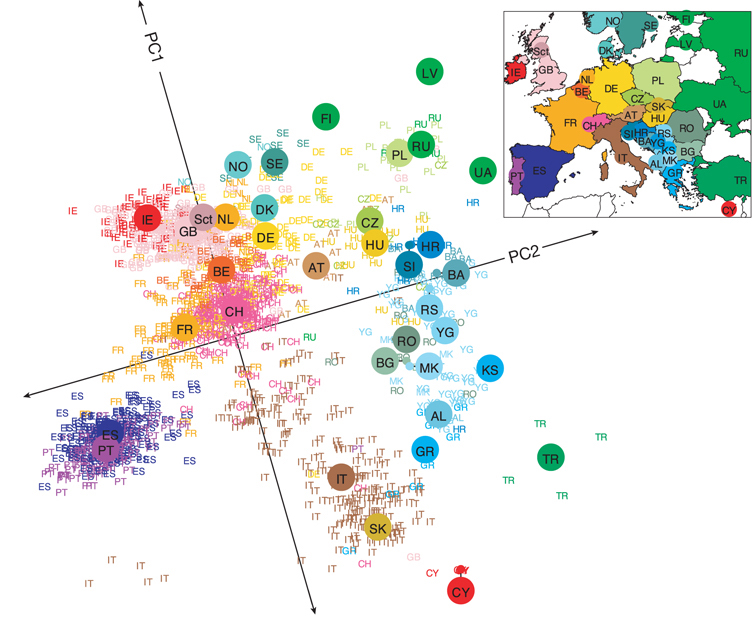
WIP: Exploring how (and when) PCA patterns recapitulate geographical history of populations, inspired by this figure by Novembre et al. (2008).
library (slendr)init_env ()
The interface to all required Python modules has been activated.
library (admixr)library (cowplot)source ("files/popgen/popgen_utils.R" )<- landscape_model (rate = 0.3 , Ne = 10000 )# pdf("exercise5.pdf", width = 8, height = 5) # for (n in c(50, 25, 10, 5, 2, 1)) { <- 10 <- landscape_sampling (model, n)<- msprime (model, samples = schedule, sequence_length = 20e6 , recombination_rate = 1e-8 ) %>% ts_mutate (1e-8 )<- ts_samples (ts)ts_eigenstrat (ts, "files/popgen/spatial_pca1" )
449 multiallelic sites (0.274% out of 163618 total) detected and removed
EIGENSTRAT object
=================
components:
ind file: files/popgen/spatial_pca1.ind
snp file: files/popgen/spatial_pca1.snp
geno file: files/popgen/spatial_pca1.geno
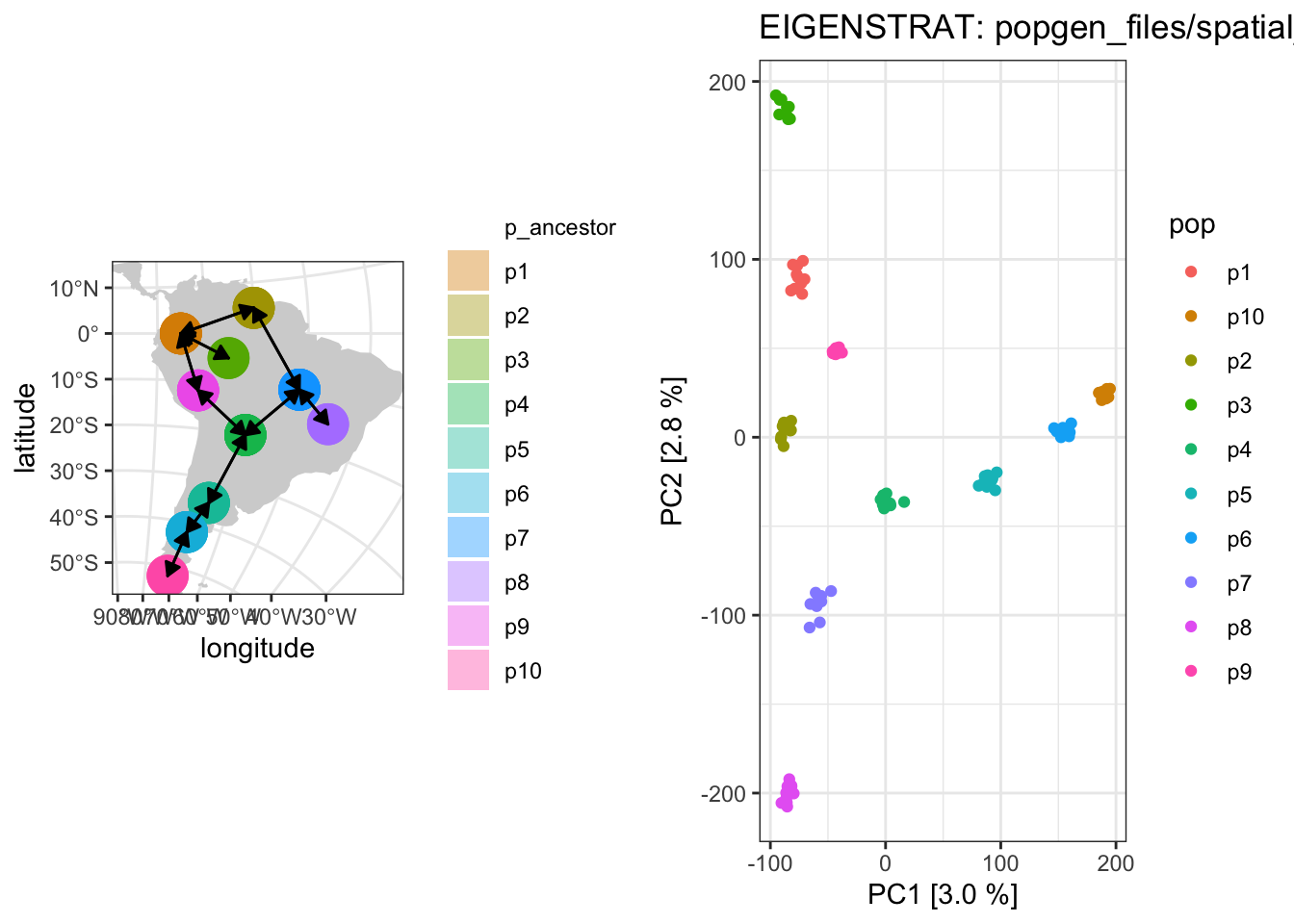
plot_pca ("files/popgen/spatial_pca1" , ts, model = "map" , color_by = "pop" )
PCA cache for the given EIGENSTRAT data was not found. Generating it now (this might take a moment)...
Warning: Visualising population labels on a map requires a higher slendr
version
Warning: All gene-flow event will be visualized at once. If you wish to visualize
gene flows at a particular point in time, use the `time` argument.
Warning: Non-spatial populations in your model won't be visualized
Per-population Ne values
library (slendr)init_env ()
The interface to all required Python modules has been activated.
library (admixr)library (cowplot)source ("files/popgen/popgen_utils.R" )<- list (p1 = 10000 ,p2 = 10000 ,p3 = 10000 ,p4 = 10000 ,p5 = 100 ,p6 = 10000 ,p7 = 10000 ,p8 = 10000 ,p9 = 10000 ,p10 = 10000 <- landscape_model (rate = 0.3 , Ne = Ne)<- list (p1 = 50 ,p2 = 50 ,p3 = 50 ,p4 = 50 ,p5 = 5 ,p6 = 50 ,p7 = 50 ,p8 = 50 ,p9 = 50 ,p10 = 50 <- landscape_sampling (model, n)<- msprime (model, samples = schedule, sequence_length = 20e6 , recombination_rate = 1e-8 ) %>% ts_mutate (1e-8 )ts_eigenstrat (ts, "files/popgen/spatial_pca2" )
1059 multiallelic sites (0.418% out of 253548 total) detected and removed
EIGENSTRAT object
=================
components:
ind file: files/popgen/spatial_pca2.ind
snp file: files/popgen/spatial_pca2.snp
geno file: files/popgen/spatial_pca2.geno
read_ind (eigenstrat ("files/popgen/spatial_pca2" ))
# A tibble: 455 × 3
id sex label
<chr> <chr> <chr>
1 p1_1 U p1_1
2 p1_2 U p1_2
3 p1_3 U p1_3
4 p1_4 U p1_4
5 p1_5 U p1_5
6 p1_6 U p1_6
7 p1_7 U p1_7
8 p1_8 U p1_8
9 p1_9 U p1_9
10 p1_10 U p1_10
# ℹ 445 more rows
# A tibble: 455 × 3
name time pop
<chr> <dbl> <chr>
1 p1_1 10000 p1
2 p1_2 10000 p1
3 p1_3 10000 p1
4 p1_4 10000 p1
5 p1_5 10000 p1
6 p1_6 10000 p1
7 p1_7 10000 p1
8 p1_8 10000 p1
9 p1_9 10000 p1
10 p1_10 10000 p1
# ℹ 445 more rows
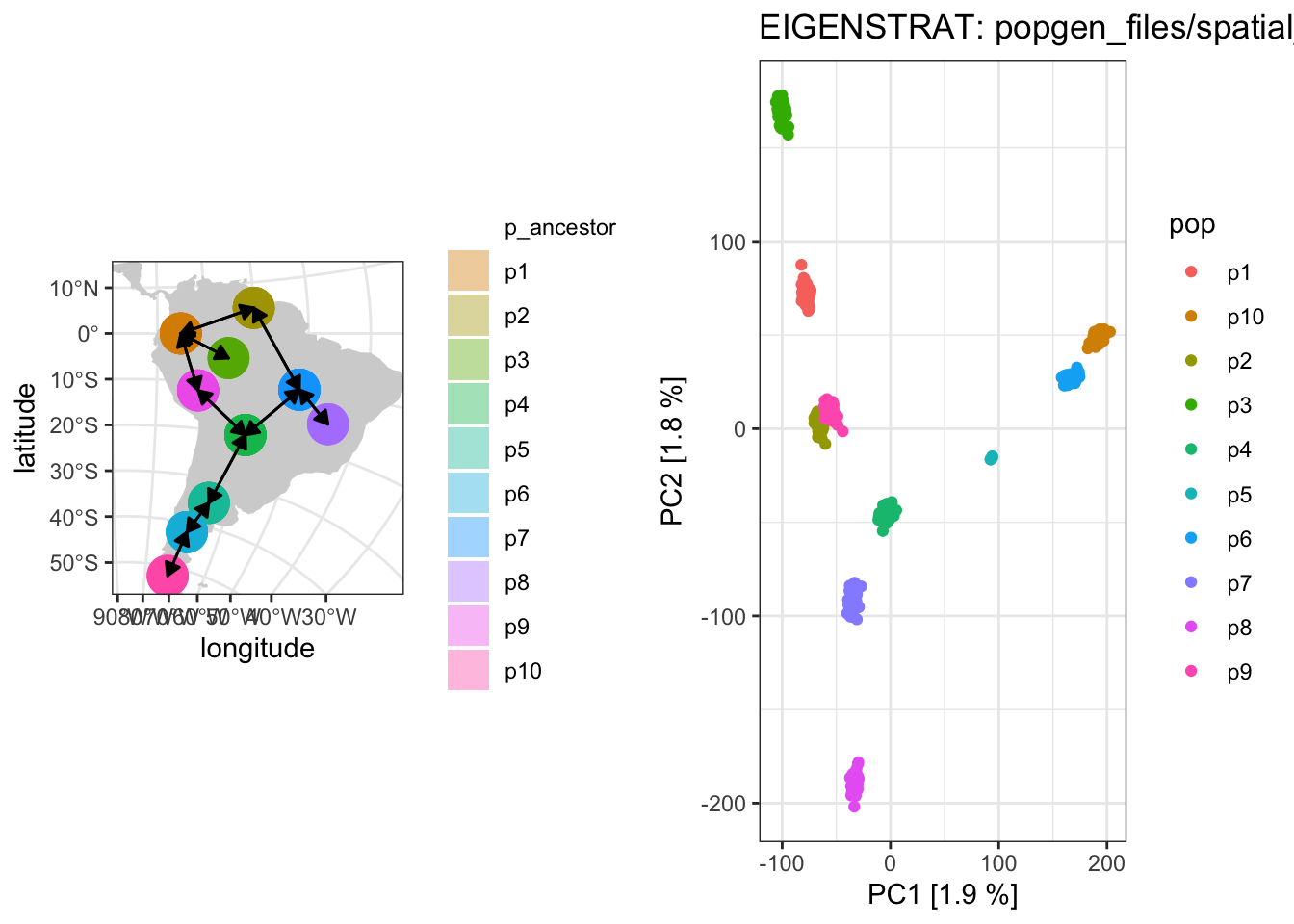
plot_pca ("files/popgen/spatial_pca2" , ts, model = "map" , color = "pop" )
PCA cache for the given EIGENSTRAT data was not found. Generating it now (this might take a moment)...
Warning: Visualising population labels on a map requires a higher slendr
version
Warning: All gene-flow event will be visualized at once. If you wish to visualize
gene flows at a particular point in time, use the `time` argument.
Warning: Non-spatial populations in your model won't be visualized