Define a gene-flow event between two populations
Usage
gene_flow(
from,
to,
start,
end,
proportion = NULL,
migration_rate = NULL,
overlap = TRUE,
rate = NULL
)Arguments
- from, to
Objects of the class
slendr_pop- start, end
Start and end of the gene-flow event
- proportion
Scalar value in the range (0, 1] specifying the total proportion of ancestry of the receiving population which will come from the source population over the entire gene-flow time window
- migration_rate
Scalar value specifying the rate of gene flow per unit of time of the given gene-flow time window
- overlap
Require spatial overlap between admixing populations? (default
TRUE)- rate
DEPRECATED (Old name for what is now the
proportionargument)
Examples
# spatial definitions -----------------------------------------------------
# create a blank abstract world 1000x1000 distance units in size
map <- world(xrange = c(0, 1000), yrange = c(0, 1000), landscape = "blank")
# create a circular population with the center of a population boundary at
# [200, 800] and a radius of 100 distance units, 1000 individuals at time 1
# occupying a map just specified
pop1 <- population("pop1", N = 1000, time = 1,
map = map, center = c(200, 800), radius = 100)
# printing a population object to a console shows a brief summary
pop1
#> slendr 'population' object
#> --------------------------
#> name: pop1
#> habitat: terrestrial
#>
#> number of spatial maps: 1
#> map: abstract spatial landscape with custom features
#> stays until the end of the simulation
#>
#> population history overview:
#> - time 1: created as an ancestral population (N = 1000)
# create another population occupying a polygon range, splitting from pop1
# at a given time point (note that specifying a map is not necessary because
# it is "inherited" from the parent)
pop2 <- population("pop2", N = 100, time = 50, parent = pop1,
polygon = list(c(100, 100), c(320, 30), c(500, 200),
c(500, 400), c(300, 450), c(100, 400)))
pop3 <- population("pop3", N = 200, time = 80, parent = pop2,
center = c(800, 800), radius = 200)
# move "pop1" to another location along a specified trajectory and saved the
# resulting object to the same variable (the number of intermediate spatial
# snapshots can be also determined automatically by leaving out the
# `snapshots = ` argument)
pop1_moved <- move(pop1, start = 100, end = 200, snapshots = 6,
trajectory = list(c(600, 820), c(800, 400), c(800, 150)))
pop1_moved
#> slendr 'population' object
#> --------------------------
#> name: pop1
#> habitat: terrestrial
#>
#> number of spatial maps: 10
#> map: abstract spatial landscape with custom features
#> stays until the end of the simulation
#>
#> population history overview:
#> - time 1: created as an ancestral population (N = 1000)
#> - time 100-200: movement across a landscape
# many slendr functions are pipe-friendly, making it possible to construct
# pipelines which construct entire history of a population
pop1 <- population("pop1", N = 1000, time = 1,
map = map, center = c(200, 800), radius = 100) %>%
move(start = 100, end = 200, snapshots = 6,
trajectory = list(c(400, 800), c(600, 700), c(800, 400), c(800, 150))) %>%
set_range(time = 300, polygon = list(
c(400, 0), c(1000, 0), c(1000, 600), c(900, 400), c(800, 250),
c(600, 100), c(500, 50))
)
# population ranges can expand by a given distance in all directions
pop2 <- expand_range(pop2, by = 200, start = 50, end = 150, snapshots = 3)
# we can check the positions of all populations interactively by plotting their
# ranges together on a single map
plot_map(pop1, pop2, pop3)
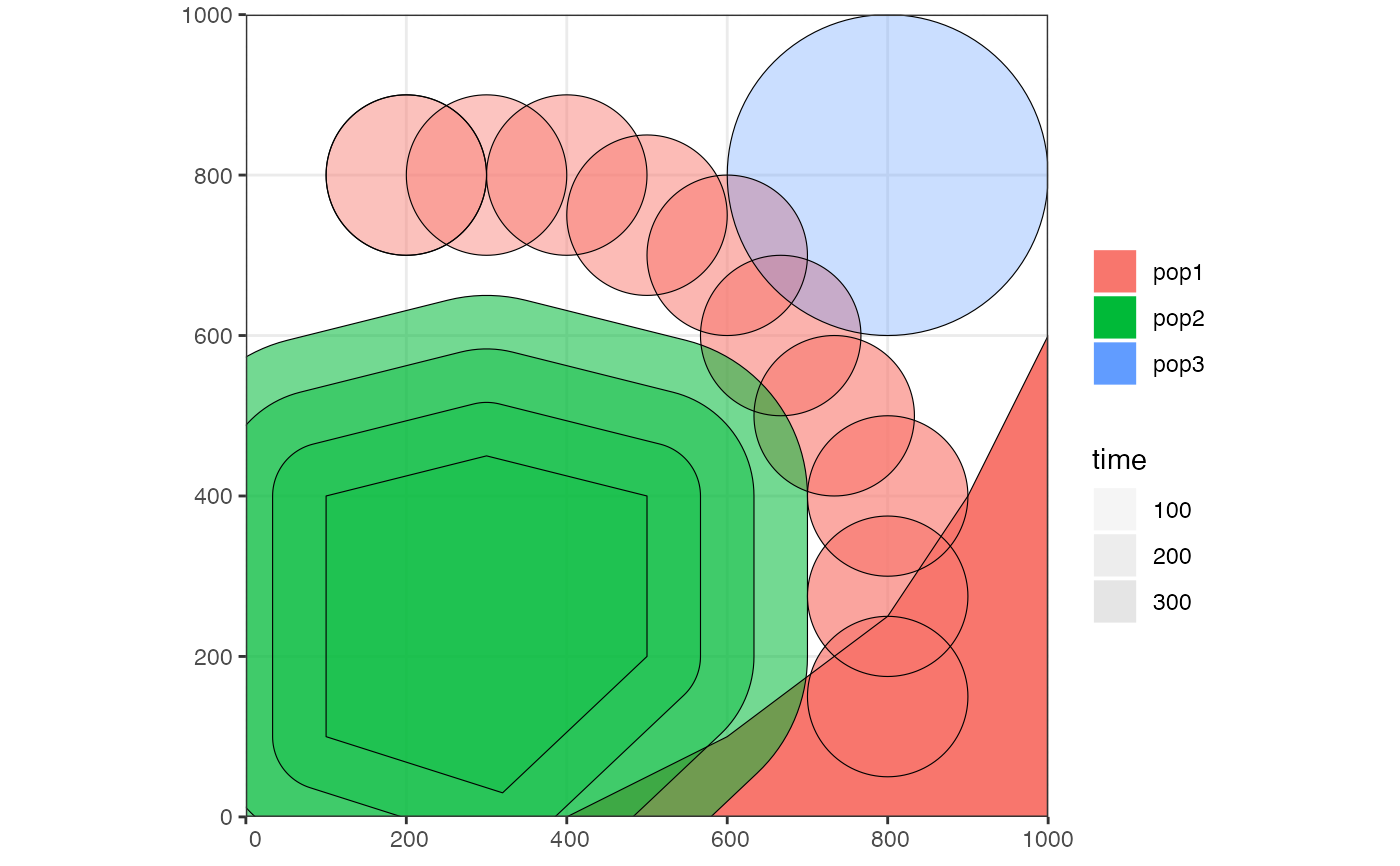 # gene flow events --------------------------------------------------------
# individual gene flow events can be saved to a list
gf <- list(
gene_flow(from = pop1, to = pop3, start = 150, end = 200, rate = 0.15),
gene_flow(from = pop1, to = pop2, start = 300, end = 330, rate = 0.25)
)
#> Warning: The argument `rate` is about to be deprecated because of its confusing
#> naming and behavior. If you want to specify the rate of migration per
#> unit of time, please use the new argument `migration_rate`. If you want
#> to specify the total amount of ancestry which the `to` population should
#> received from the `from` population, use the new argument `proportion`
#> (this corresponds to the original interpretation of the deprecated `rate`
#> argument, and a simple replacement of `rate` with `proportion` will thus
#> retain the original meaning of your code all).
#> Warning: The argument `rate` is about to be deprecated because of its confusing
#> naming and behavior. If you want to specify the rate of migration per
#> unit of time, please use the new argument `migration_rate`. If you want
#> to specify the total amount of ancestry which the `to` population should
#> received from the `from` population, use the new argument `proportion`
#> (this corresponds to the original interpretation of the deprecated `rate`
#> argument, and a simple replacement of `rate` with `proportion` will thus
#> retain the original meaning of your code all).
# compilation -------------------------------------------------------------
# compile model components in a serialized form to dist, returning a single
# slendr model object (in practice, the resolution should be smaller)
model <- compile_model(
populations = list(pop1, pop2, pop3), generation_time = 1,
resolution = 100, simulation_length = 500,
competition = 5, mating = 5, dispersal = 1
)
# gene flow events --------------------------------------------------------
# individual gene flow events can be saved to a list
gf <- list(
gene_flow(from = pop1, to = pop3, start = 150, end = 200, rate = 0.15),
gene_flow(from = pop1, to = pop2, start = 300, end = 330, rate = 0.25)
)
#> Warning: The argument `rate` is about to be deprecated because of its confusing
#> naming and behavior. If you want to specify the rate of migration per
#> unit of time, please use the new argument `migration_rate`. If you want
#> to specify the total amount of ancestry which the `to` population should
#> received from the `from` population, use the new argument `proportion`
#> (this corresponds to the original interpretation of the deprecated `rate`
#> argument, and a simple replacement of `rate` with `proportion` will thus
#> retain the original meaning of your code all).
#> Warning: The argument `rate` is about to be deprecated because of its confusing
#> naming and behavior. If you want to specify the rate of migration per
#> unit of time, please use the new argument `migration_rate`. If you want
#> to specify the total amount of ancestry which the `to` population should
#> received from the `from` population, use the new argument `proportion`
#> (this corresponds to the original interpretation of the deprecated `rate`
#> argument, and a simple replacement of `rate` with `proportion` will thus
#> retain the original meaning of your code all).
# compilation -------------------------------------------------------------
# compile model components in a serialized form to dist, returning a single
# slendr model object (in practice, the resolution should be smaller)
model <- compile_model(
populations = list(pop1, pop2, pop3), generation_time = 1,
resolution = 100, simulation_length = 500,
competition = 5, mating = 5, dispersal = 1
)